
Chapter 6 Clustering
At its core, clustering is a tool that allows us to examine structure and patterns in our data. There isn’t really a “true” answer to how the data should be clustered. Instead, we can change the algorithms and parameters to explore a variety of possibilities that work best for each dataset and question the researcher is trying to answer.
6.1 Clustering using graph-based methods
Graph-based clustering is based on identifying the nearest neighbors of each cell in high-dimensional space. The connections between a cell and its neighbors (called edges) are weighted based on the similarity of the two cells connected. An edge is assigned a higher weight if the two cells it connects are more closely related. After all cells have been connected to their neighbors, we apply an algorithm to identify clusters, or communities, of related cells. Each cell within a community will be more closely related to any cell within the same community than to cells outside the community.
Graph-based clustering scales easily, because it only used a k-nearest neighbor search. These searches run more quickly than other methods (like hierarchical clustering). Unfortunately, no information is retained about relationships beyond the neighboring cells. This effect also means that clustering resolution depends on cell density.
# let the R algorithm define and label our clusters
nn.clusters <- clusterCells(sce.zeisel.tsne20, use.dimred="PCA")
# this command tells us how many clusters were identified (the top row) and how many cells belong to each cluster (the bottom row)
table(nn.clusters)## nn.clusters
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
## 284 147 107 195 604 166 475 62 119 256 221 67 53 28 32We assigned the cluster assignments back into our SingleCellExperiment object as a factor in the column metadata, which allows us to visualize the cluster assignment on a t-SNE plot.
# create a t-SNE plot showing the identified clusters
colLabels(sce.zeisel.tsne20) <- nn.clusters
plotReducedDim(sce.zeisel.tsne20, "TSNE", colour_by="label")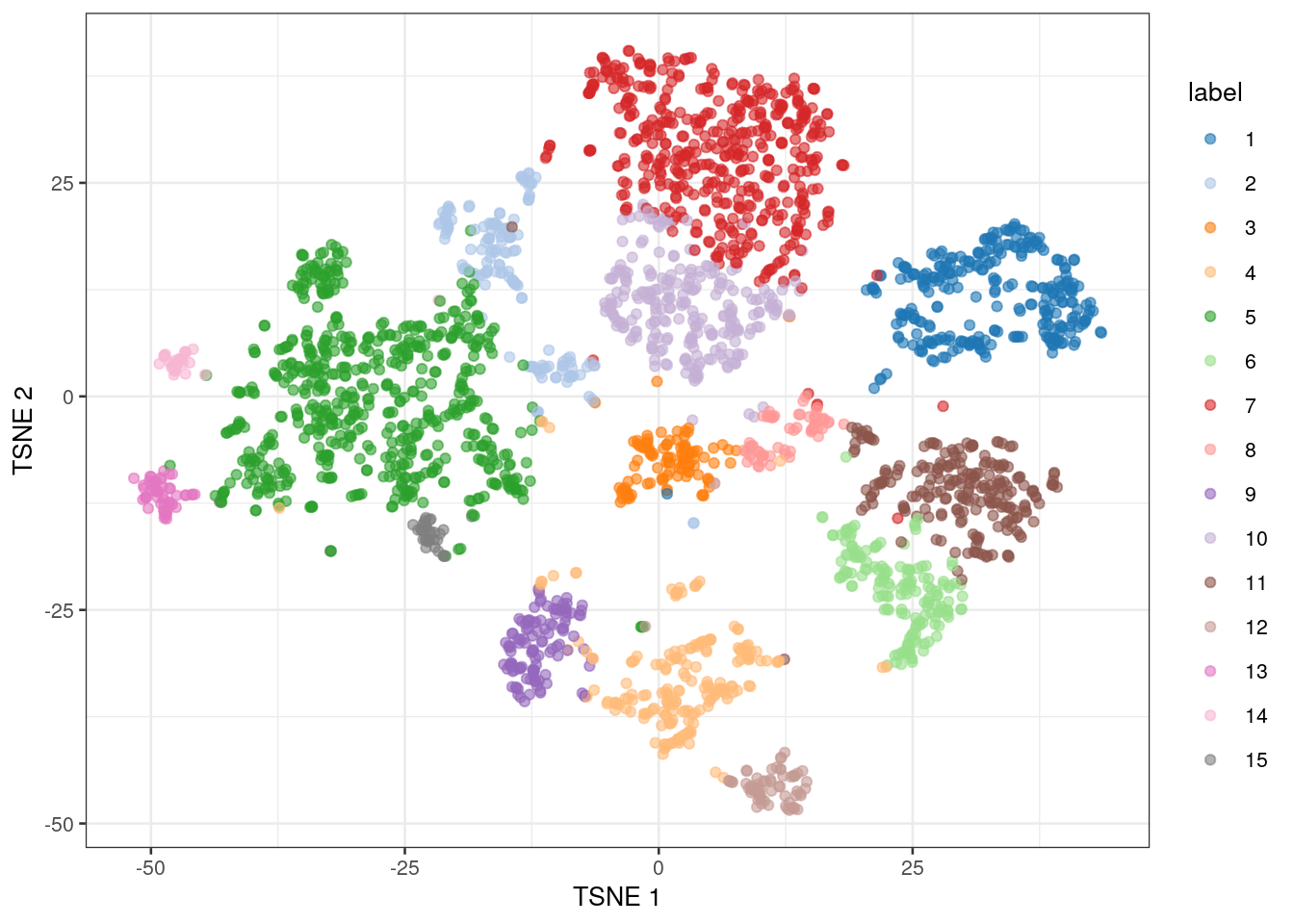
QUESTIONS 1. How many clusters were identified using graph-based clustering? Which cluster contained the most cells, and how many cells did it have?
sessionInfo()## R version 4.1.3 (2022-03-10)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 20.04.5 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/liblapack.so.3
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] uwot_0.1.11 Matrix_1.4-0
## [3] BiocSingular_1.10.0 scran_1.22.1
## [5] scater_1.22.0 ggplot2_3.3.5
## [7] scuttle_1.4.0 scRNAseq_2.8.0
## [9] SingleCellExperiment_1.16.0 SummarizedExperiment_1.24.0
## [11] Biobase_2.54.0 GenomicRanges_1.46.1
## [13] GenomeInfoDb_1.30.1 IRanges_2.28.0
## [15] S4Vectors_0.32.4 BiocGenerics_0.40.0
## [17] MatrixGenerics_1.6.0 matrixStats_0.61.0
##
## loaded via a namespace (and not attached):
## [1] AnnotationHub_3.2.2 BiocFileCache_2.2.1
## [3] igraph_1.3.1 lazyeval_0.2.2
## [5] BiocParallel_1.28.3 digest_0.6.29
## [7] ensembldb_2.18.4 htmltools_0.5.2
## [9] viridis_0.6.2 fansi_1.0.3
## [11] magrittr_2.0.3 memoise_2.0.1
## [13] ScaledMatrix_1.2.0 cluster_2.1.2
## [15] limma_3.50.3 Biostrings_2.62.0
## [17] prettyunits_1.1.1 colorspace_2.0-3
## [19] blob_1.2.3 rappdirs_0.3.3
## [21] ggrepel_0.9.1 xfun_0.26
## [23] dplyr_1.0.8 crayon_1.5.1
## [25] RCurl_1.98-1.6 jsonlite_1.8.0
## [27] glue_1.6.2 gtable_0.3.0
## [29] zlibbioc_1.40.0 XVector_0.34.0
## [31] DelayedArray_0.20.0 scales_1.2.1
## [33] DBI_1.1.2 edgeR_3.36.0
## [35] Rcpp_1.0.8.3 viridisLite_0.4.0
## [37] xtable_1.8-4 progress_1.2.2
## [39] dqrng_0.3.0 bit_4.0.4
## [41] rsvd_1.0.5 metapod_1.2.0
## [43] httr_1.4.2 FNN_1.1.3
## [45] ellipsis_0.3.2 farver_2.1.0
## [47] pkgconfig_2.0.3 XML_3.99-0.9
## [49] sass_0.4.1 dbplyr_2.1.1
## [51] locfit_1.5-9.5 utf8_1.2.2
## [53] labeling_0.4.2 tidyselect_1.1.2
## [55] rlang_1.0.2 later_1.3.0
## [57] AnnotationDbi_1.56.2 munsell_0.5.0
## [59] BiocVersion_3.14.0 tools_4.1.3
## [61] cachem_1.0.6 cli_3.2.0
## [63] generics_0.1.2 RSQLite_2.2.12
## [65] ExperimentHub_2.2.1 evaluate_0.15
## [67] stringr_1.4.0 fastmap_1.1.0
## [69] yaml_2.3.5 knitr_1.33
## [71] bit64_4.0.5 purrr_0.3.4
## [73] KEGGREST_1.34.0 AnnotationFilter_1.18.0
## [75] sparseMatrixStats_1.6.0 mime_0.12
## [77] xml2_1.3.3 biomaRt_2.50.3
## [79] compiler_4.1.3 beeswarm_0.4.0
## [81] filelock_1.0.2 curl_4.3.2
## [83] png_0.1-7 interactiveDisplayBase_1.32.0
## [85] statmod_1.4.36 tibble_3.1.6
## [87] bslib_0.3.1 stringi_1.7.6
## [89] highr_0.9 RSpectra_0.16-0
## [91] GenomicFeatures_1.46.5 lattice_0.20-45
## [93] bluster_1.4.0 ProtGenerics_1.26.0
## [95] vctrs_0.4.1 pillar_1.7.0
## [97] lifecycle_1.0.1 BiocManager_1.30.16
## [99] jquerylib_0.1.4 BiocNeighbors_1.12.0
## [101] bitops_1.0-7 irlba_2.3.5
## [103] httpuv_1.6.5 rtracklayer_1.54.0
## [105] R6_2.5.1 BiocIO_1.4.0
## [107] bookdown_0.24 promises_1.2.0.1
## [109] gridExtra_2.3 vipor_0.4.5
## [111] assertthat_0.2.1 rjson_0.2.21
## [113] withr_2.5.0 GenomicAlignments_1.30.0
## [115] Rsamtools_2.10.0 GenomeInfoDbData_1.2.7
## [117] parallel_4.1.3 hms_1.1.1
## [119] grid_4.1.3 beachmat_2.10.0
## [121] rmarkdown_2.10 DelayedMatrixStats_1.16.0
## [123] Rtsne_0.16 shiny_1.7.1
## [125] ggbeeswarm_0.6.0 restfulr_0.0.13