
Chapter 4 Calculate idxstats on multiple files
Now that you’ve successfully written an introductory WDL, this chapter demonstrates how WDL Workflows can easily perform an analysis across multiple genomic data files. This material is adapted from the WDL Bootcamp workshop. For more hands-on WDL-writing exercises, see Hands-on practice for scripting and configuring Terra workflows.
Learning Objectives
- Solidify your understanding of a WDL script’s structure
- Modify a template to write a more complex WDL
- Understand how to customize a workflow’s setup with WDL
4.1 Clone data workspace
Let’s start by navigating to the demos-combine-data-workspaces on AnVIL-powered-by-Terra.
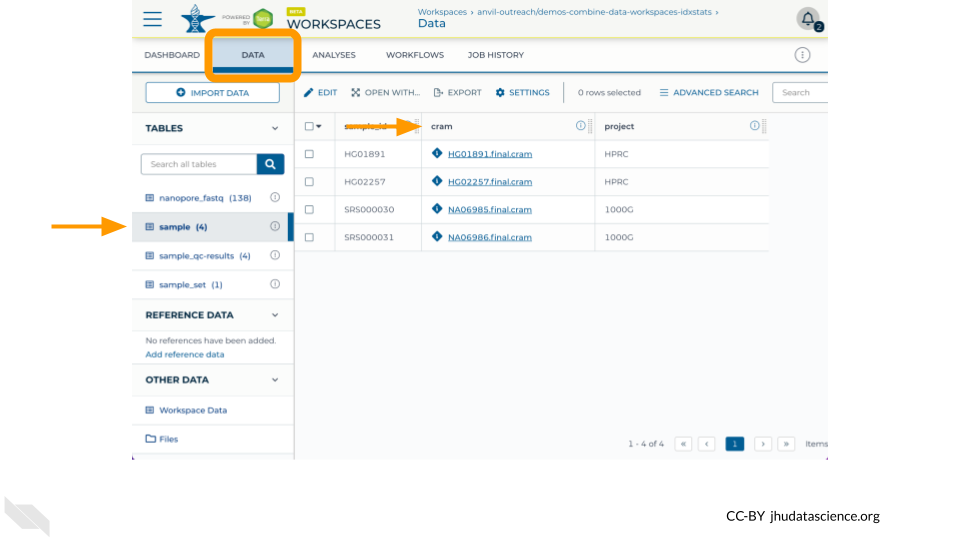
This workspace contains a Data Table named sample which contains references to four .cram files, two from the 1000 Genomes Project and two from the Human Pangenome Reference Consortium.
Clone this workspace to create a place where you can organize your analysis.
Examine the sample table in the Data tab to ensure that you see references to four .cram files.
In the next steps, you will write a WDL to analyze these .cram files, run the Workflow, and examine the output.
4.2 Write an idxstats WDL
To build off of your hello-input WDL, let’s practice writing a more complex WDL. In this exercise, you’ll fill in a template script to calculate Quality Control (QC) metrics for a BAM/CRAM file using the samtools idxstats function. For this exercise, there are two WDL runtime parameters that we must update for the Workflow to succeed:
docker– Specify a Docker image that contains necessary softwaredisks– Increase the disk size for each provisioned resource
Start by downloading the template script shown below. Open it in a text editor and modify it to call samtools idxstats on a bam file.
version 1.0
workflow samtoolsIdxstats {
input {
File bamfile
}
call {
input:
}
output {
}
}
task {
input {
}
command <<<
>>>
output {
}
runtime {
docker: ''
}
}Importantly, you must:
- Specify a Docker image that contains SAMtools (e.g.
ekiernan/wdl-101:v1) - Increase the disk size for each provisioned resource, e.g.
local-disk 50 HDD
Hints:
- Follow the same general method as in the “Hello World” exercise in section 3.3.
- In this case, the input will be a BAM file.
- The output will be a file called
idxststats.txt. - The task will be to calculate QC metrics, using this command:
samtools index ~{bamfile}
samtools idxstats ~{bamfile} > counts.txtThen, compare your version to the completed version below:
version 1.0
workflow samtoolsIdxstats {
input {
File bamfile
}
call idxstats {
input:
bamfile = bamfile
}
output {
File results = idxstats.idxstats
}
}
task idxstats {
input {
File bamfile
}
command <<<
samtools index ~{bamfile}
samtools idxstats ~{bamfile} > idxstats.txt
>>>
output {
File idxstats = "idxstats.txt"
}
runtime {
disks: 'local-disk 50 HDD'
docker: 'ekiernan/wdl-101:v1'
}
}4.3 Optional: Run idxstats WDL on multiple .bam files
You can test this workflow out by creating a new method in the Broad Methods Repository, exporting it to your clone of the demos-combine-data-workspaces workspace, and running it on samples in the ’sample data table.
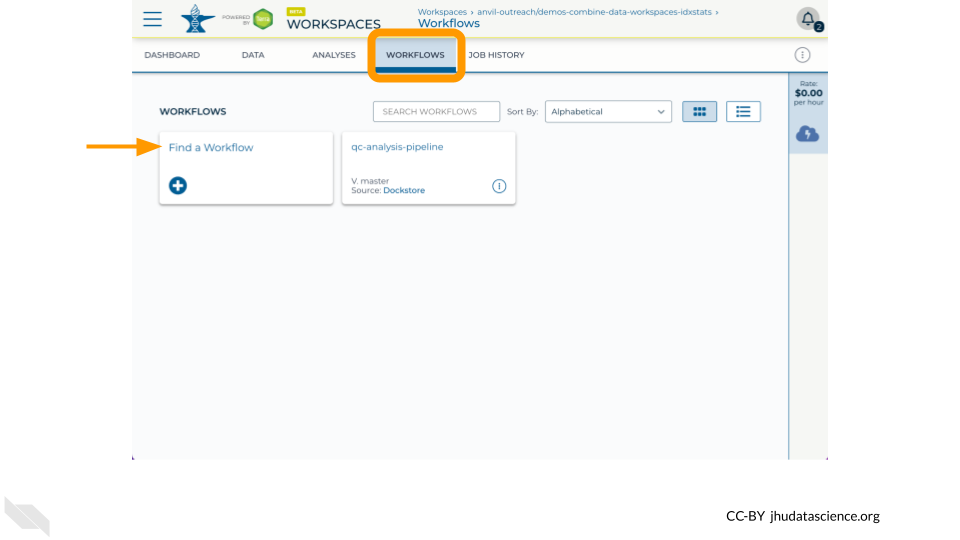
First, go to the “Workflows” tab and access the Broad Methods Repository through the “Find a Workflow” card:
Copy and paste the idxstats WDL you wrote above and export to your workspace (see Chapter 3 if you need a refresher). Next, select “Run workflow(s) with inputs defined by data table” and choose the .cram files that you wish to analyze:
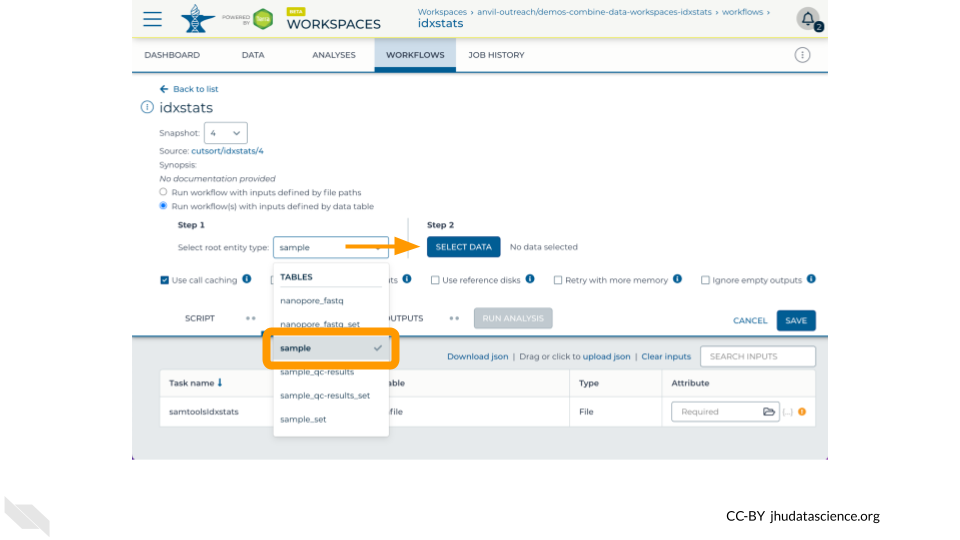
- Step 1: Select the
sampletable in the root entity type drop-down menu - Step 2: Click “Select Data” and tick the checkboxes for one or more rows in the data table
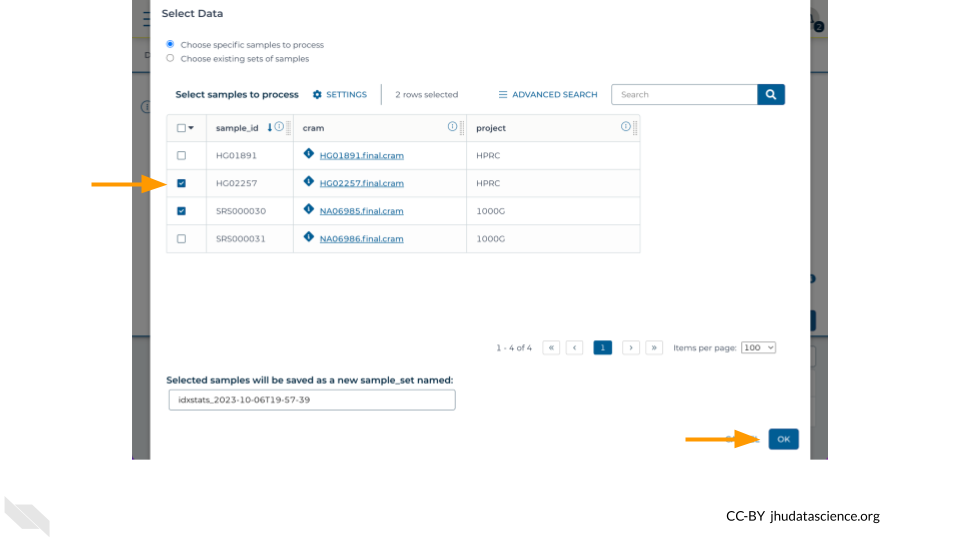
Finally, configure the “Inputs” tab by specifying this.cram as the Attribute for the variable bamfile for the task samtoolsIdxstats. Don’t forget to click “Save”.
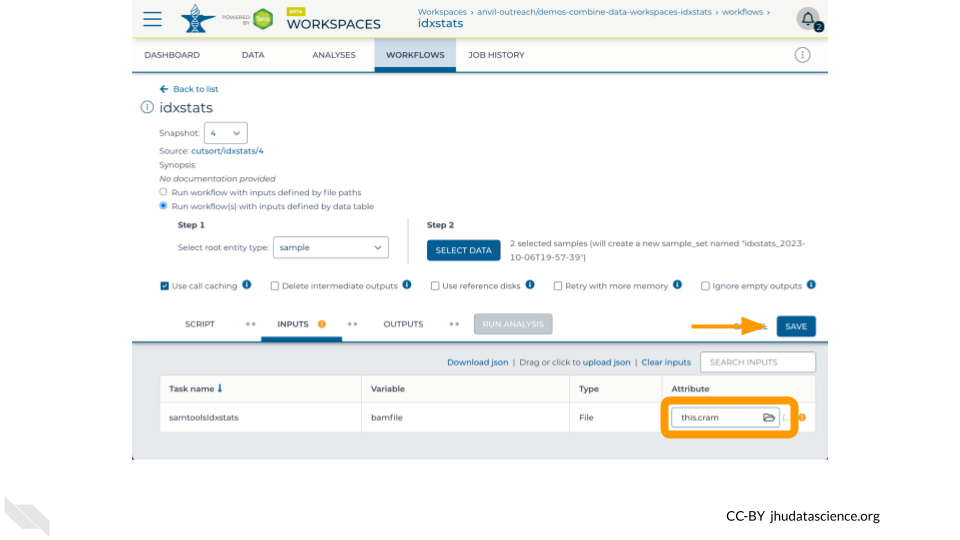
Now run the job by clicking “Run Analysis”! You can monitor the progress from “Queued” to “Running” to “Succeeded” in the “Job History” tab
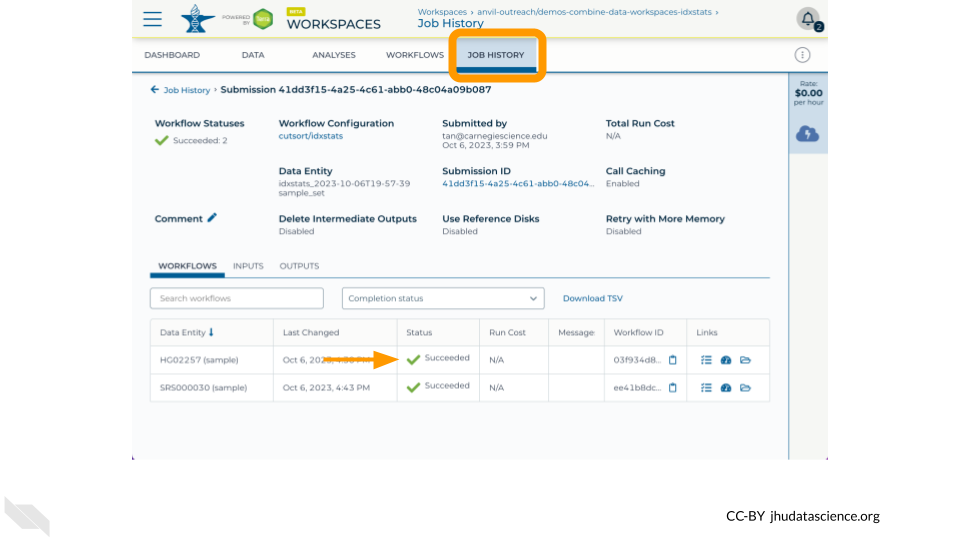
Once the job is complete, navigate to the “Data” tab and click on “Files” to find the idxstats.txt output and logs by traversing through
submissions/<submission_id>/samtoolsIdxstats/<workflow_id>/call-idxstats/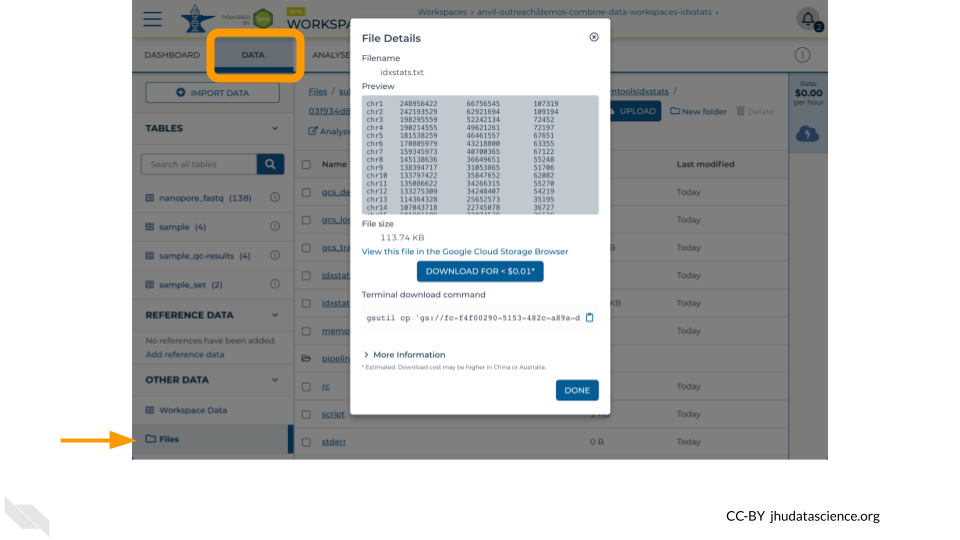
4.4 Customize your Workflow’s Setup with WDL
In addition to defining the workflow’s tasks, WDL scripts can define how your workflow runs in AnVIL-powered-by-Terra.
4.4.1 Memory retry
Some workflows require more memory than others. But, memory is not free, so you don’t want to request more memory than you need. One solution to this tension is to start with a small amount of memory and then request more if you run out of memory. Learn how to do this from your WDL script by reading Out of Memory Retry, and see Workflow setup: VM and other options for a general overview of how to set up your workflow’s compute resources.
4.4.2 Localizing files
It can be hard to know where your data files are located within your workspace bucket – the folders aren’t intuitively named, and often your files are saved several folders deep.
Luckily, WDL scripts can localize your files for you. For more on this, see How to configure workflow inputs, How to use DRS URIs in a workflow, and Creating a list file of reads for input to a workflow.
If your workflow generates files, you can also write their location to a data table. This is useful for both intermediate files and the workflow’s final outputs. For more on this topic, see Writing workflow outputs to the data table.